От того, что вы потребуете всегда один и тот же знак 3-ей производной, ни лучше, не хуже не будет. Вы же все равно минимизируете функцию суммы. Вот на нее и смотреть надо, а вот у функций по торсионным и по обычным углам третья производная должна менять знак, так как это функция от угла, который в первом случае в 2Пи, а во втором случае, в Пи вписывается, и вот тут нужны совокупные характеристики функционала, иначе то, что вы будете минимизировать будет иметь кучу оврагов, минимизатор на них будет садиться, или даже вообще в них застревать, и у вас будет реально лажа.
Я говорю, возьмите K(r-r0)^2 на все связи, углы и торсионные углы, добавьте простейшего ван-дер-ваальса, и GSLем пооптимизируйте и на результаты посмотрите. За вечер это можно запрограммировать. Реально будет больше пользы, чем абстрактно филосовствовать о третьей производной, зато получите бесценный опыт и начнете понимать почему там все там интересно.
А по-хорошему, без пары книг по методам минимизации, вдоль и поперек прочитанных и понятых, попытки запрограммиорвать что-то будут похожи на ходьбу по минному полю - шаг влево-вправо от на первый взгляд странно выведенной формулы в чужой статье будет приводить к совершенно не трактуемым результатам.
Можно кстати, начать с хабра, например, отсюда:
https://habr.com/ru/articles/470638/
https://habr.com/ru/articles/561128/
а из книг рекомендую
Nocedal, J., Wright, S. J., Numerical Optimization. Second Edition. Springer 2006.
Kelley, C. T. (1999), Iterative Methods for Optimization, Philadelphia: Society for Industrial and Applied Mathematics.
ну и википедию на тему
https://en.wikipedia.org/wiki/Broyden%E ... _algorithm
Реализация ab initio подхода в молекулярной механике

Re: Реализация ab initio подхода в молекулярной механике
Я готов взяться за эту задачу, хотя уйдёт побольше дня. Но хотелось бы вначале определиться с теорией, чтобы не слишком много потом пришлось переделывать.
Повторю свой вопрос - что считать "фундаментальными принципами" для молекулярной механики. Очевидно всё сводится к электромагнитным взаимодействиям. Уточню - электростатическим или именно электромагнитным? В молекулах магнетизм играет такую же важную роль? Если не путаю, магнетизм это следствие релятивизма?
У меня создалось ощущение, что мы до сих пор толком не понимаем, что такое электромагнитные (даже может быть только электростатические) взаимодействия. Попробую объяснить. Вначале мы узнали, что такое электростатические (кулоновские) силы, взаимодействие заряженных частиц. Потом оказывается, что есть диполь-дипольное взаимодействие. Потом оказывается, что есть взаимодействие наведённых диполей, ориентационное, индукционное (я уже не очень помню что это такое конкретно). Потом оказывается, что есть взаимодействие в куперовских парах. Есть ли этому предел? Не знаю, понятно ли я сформулировал.
Повторю свой вопрос - что считать "фундаментальными принципами" для молекулярной механики. Очевидно всё сводится к электромагнитным взаимодействиям. Уточню - электростатическим или именно электромагнитным? В молекулах магнетизм играет такую же важную роль? Если не путаю, магнетизм это следствие релятивизма?
У меня создалось ощущение, что мы до сих пор толком не понимаем, что такое электромагнитные (даже может быть только электростатические) взаимодействия. Попробую объяснить. Вначале мы узнали, что такое электростатические (кулоновские) силы, взаимодействие заряженных частиц. Потом оказывается, что есть диполь-дипольное взаимодействие. Потом оказывается, что есть взаимодействие наведённых диполей, ориентационное, индукционное (я уже не очень помню что это такое конкретно). Потом оказывается, что есть взаимодействие в куперовских парах. Есть ли этому предел? Не знаю, понятно ли я сформулировал.
Re: Реализация ab initio подхода в молекулярной механике
Уважаемый Vit Nhoc,
давайте вспомним, что в этой теме вы пытаетесь научиться азам молекулярной механики и уважаемые специалисты в лице Гесса, madschumacher очень много Вам рассказали, и я тоже много набросил.
Я конечно рад, что задачку уровня университетского практикума второго курса по Numeruk II вы все-таки планируете осилить, но, опять же, вспомните, это Вам надо, не мне, и не Гессу. У меня есть 1.7 миллиардов конформеров посчитанных моим софтом, и куча немецого университетского народу сейчас это успешно обсуждает.
Вам же здесь в этой теме преподают азы, чтобы Вы быстрее въехали в тему, не забывая рассказывать о куче грабль, которые там распиханы и которые надо обходить со знанием дела, а от Вас даже спасибо не видно.
Re: Реализация ab initio подхода в молекулярной механике
Chemigor, я потом почитаю ваши ссылки по методам градиентного спуска, но это далеко не главное. У меня довольно большой опыт разных алгоритмов оптимизации чего-нибудь, я это могу реализовать и без подсказок, только вначале у меня алгоритм оптимизации будет медленный (зато надёжный - всегда коробит когда в оптимизации Оркой или Гауссианом какая-то из геометрий имеет энергию чуть ниже последней, у меня бы такого никогда не было). Но сейчас очень интересно обсудить теоретические принципы. Я имею достаточно большой опыт расчётов разных соединений (хотя обычно я это не публикую, лень писать статью), разных классов, и моя интуиция, натренированная на этой выборке, говорит что модель, основанная только на притягивании и отталкивании атомов и фрагментов, должна работать лучше, чем модель с валентными углами, а тем более двугранными.

Re: Реализация ab initio подхода в молекулярной механике
Можно чисто практический вопрос? Как вы оцениваете, сколько времени вам потребовалось бы, чтобы накодить экспериментальный образец такой модели, самый простейший: С(sp3), О(sp3), Н? Т.е. только алканы, простые эфиры, спирты и т.д. Никакой ароматики, непредельщины и карбонилов.
Когда начинает изменять память, практики заводят записную книжку, а романтики садятся писать мемуары.
Re: Реализация ab initio подхода в молекулярной механике
Думаю, от одной до двух недель, если алгоритм оптимизации энергии будет медленный. Когда возьмусь, могу посчитать время и отчитаться вам. Но пока я хочу определиться с теорией, чтобы не переделывать потом всё.Ahha писал(а): ↑Сб авг 12, 2023 8:47 pmМожно чисто практический вопрос? Как вы оцениваете, сколько времени вам потребовалось бы, чтобы накодить экспериментальный образец такой модели, самый простейший: С(sp3), О(sp3), Н? Т.е. только алканы, простые эфиры, спирты и т.д. Никакой ароматики, непредельщины и карбонилов.
Re: Реализация ab initio подхода в молекулярной механике
Магнитное поле вполне описывается уравнениями Максвелла, которые классические.
Нет, передо мной отчитываться не нужно: я вам не начальник и не грантодатель.
Когда начинает изменять память, практики заводят записную книжку, а романтики садятся писать мемуары.

Re: Реализация ab initio подхода в молекулярной механике
При расчете на RDFT вы получите гетеролиз связи, разрыв на катион и анион. Не совсем то что вам надо но тоже результат.
Я не в курсе есть ли форсфилды описывающие магнетизм ака спиновые состояния. ИМХО нет, ибо тогда бы они где нибудь проскакисвали в металлокомплексах и я бы это видел в каких нибудь статьях.
Ну я попробовал выше сформулировать почему на мой взгляд это не сработает. Но разумеется можете попробовать.
Вариантов на самом деле не так много:
1) это действительно рабочий вариант и он используется, но я тупо о нем не в курсе (что возможно ибо... сколько раз я уже сказал что форсфилды это не мое поле?)
2) это не работает, например по причинам которые я озвучил или по каким то другим (я нахожусь здесь).
3) это рабочая идея до которой не додумались десятки профессоров и сотни постдоков которые как минимум с 1974ого и до сегодня изобрели несколько десятков силовых полей. Не то чтобы это было принципиально невозможно, но мне кажется это маловероятным.
С одной стороны по пункту 1 вам бы стоило пообщаться с кем то из экспертов, с другой стороны если не 1 - то я думаю эксперты возьмутся за отстаивание версии "невозможно" намного интенсивнее чем я.
Re: Реализация ab initio подхода в молекулярной механике
я попрыгал по литературе, я не вижу никаких связок магнетизма и форсфилдов.
Re: Реализация ab initio подхода в молекулярной механике
Релятивистские эффекты работают не в уравнениях, а в природе. Релятивизм - не причина магнетизма, а просто следующее приближение к построению полной картины мира. Уравнения Максвелла были выведены исходя из классических представлений, но в релятивизм вписались отлично, да.
Не сочтите за резкость, но лично мне начинает казаться, что таким "определением с теорией" вы через достаточно продолжительное время дойдете до несложного вывода:
Когда начинает изменять память, практики заводят записную книжку, а романтики садятся писать мемуары.
Re: Реализация ab initio подхода в молекулярной механике
Ну у меня на самом деле очень простой вопрос: правильно ли я понимаю, что для лёгких элементов, например простой органики, релятивистские эффекты не особо важны? Если например скорость света увеличится в три раза, на рассчитанные свойства фенола это почти не отразится?Ahha писал(а): ↑Вс авг 13, 2023 4:47 pmРелятивистские эффекты работают не в уравнениях, а в природе. Релятивизм - не причина магнетизма, а просто следующее приближение к построению полной картины мира. Уравнения Максвелла были выведены исходя из классических представлений, но в релятивизм вписались отлично, да.
Re: Реализация ab initio подхода в молекулярной механике
Да, даже в квантах до условно говоря брома релятивизмом народ не заморачивается никогда, а смелые и бедные и йод и золото считают без релятивистских поправок.
Но магнитные свойства отлично наличествуют уже у 3d металлов где релятивизмом еще и рядом не пахнет.
-
madschumacher

- Сообщения: 883
- Зарегистрирован: Ср авг 05, 2015 4:30 pm
Re: Реализация ab initio подхода в молекулярной механике
Даже хуже, магнитные свойства есть уже у кислорода, которым мы дышим, O2 то бишь. Вполне себе парамагнетик.
И да узрел Охламон, что сие есть круть несусветная!
Re: Реализация ab initio подхода в молекулярной механике
А вот молмеху они нужны?madschumacher писал(а): ↑Пн авг 14, 2023 12:17 amДаже хуже, магнитные свойства есть уже у кислорода, которым мы дышим, O2 то бишь. Вполне себе парамагнетик.
Ну будет у нас кислород без магнитного момента (аля синглет), с длиной пружинки как в триплете. И кому хуже?
Re: Реализация ab initio подхода в молекулярной механике
У меня вопрос: как химики объясняют вот такие связи?
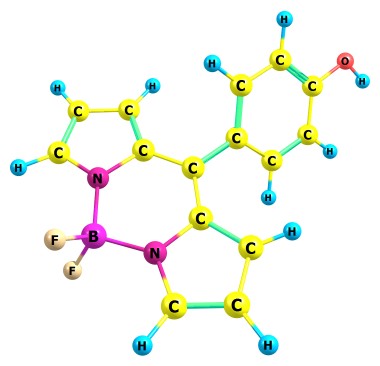
Если не ошибаюсь, это общая история для порфиринов, точнее для металлопорфиринов.
Если не ошибаюсь, это общая история для порфиринов, точнее для металлопорфиринов.
Re: Реализация ab initio подхода в молекулярной механике
в начале 90-х на семинаре то ли у Пупышева, то ли у Степанова, то ли их обоих, как-то обсуждалась какая-то публикация, когда мол-мех был записан в виде только притягивания-отталкивания всех атомов с разными весами и формулами. Преподносилось это как альтернатива вычислению углов, а именно арккосинуса, который в те давние времена на большинстве персоналок считался реально долго. Особенно тогда педалировалась идея с торсионными углами - де, чтобы их вычислить надо считать несколько раз углы и проекции, а тут раз, и только один корень для расстояния.
Как я помню результаты были хуже, но кого-то это не останавливало.
Потом, как я понимаю, идея заглохла, так как люди научились считать те же тригонометрические функции кордиком и быстрее, и все проблемы просто рассосались.
Re: Реализация ab initio подхода в молекулярной механике
Мне кажется вы что-то путаете, проблема с длительностью считания арккосинуса актуальна только с деревянными счётами.chemigor писал(а): ↑Пн авг 14, 2023 5:08 pmв начале 90-х на семинаре то ли у Пупышева, то ли у Степанова, то ли их обоих, как-то обсуждалась какая-то публикация, когда мол-мех был записан в виде только притягивания-отталкивания всех атомов с разными весами и формулами. Преподносилось это как альтернатива вычислению углов, а именно арккосинуса, который в те давние времена на большинстве персоналок считался реально долго. Особенно тогда педалировалась идея с торсионными углами - де, чтобы их вычислить надо считать несколько раз углы и проекции, а тут раз, и только один корень для расстояния.
Как я помню результаты были хуже, но кого-то это не останавливало.
Потом, как я понимаю, идея заглохла, так как люди научились считать те же тригонометрические функции кордиком и быстрее, и все проблемы просто рассосались.
Re: Реализация ab initio подхода в молекулярной механике
Я даже не знаю, что и ответить, в 90-е годы, про которые я говорил, большинство софта для молмеха писали в целочисленной арифметике, так как не у каждого компьютера был сопроцессор, и если расстояние между атомами в квадрате считалось на раз, вот на всякой тригонометрии ужасно садилась производительность.
Если вы в то время не жили и активно не разрабатывали вычислительные решатели, могли бы хотя бы погуглить, чем беспочвенно бросаться обвинениями.
В современном мире это конечно не так, арккосинусы только в 50 раз медленнее обычных хорошо распараллеленных умножений, поэтому я полностью соглашусь со всеми тремя пунктами, что перечислил Гесс.
Re: Реализация ab initio подхода в молекулярной механике
Это bodipy.
Ну по большому счету ничего сверхестественного, какое нибудь NBO наверное классифицирует одну связь B-N как обычную ковалентную, вторую как доноракцепторную, соответственно у одного пиррольного кольца связь с центральным углеродом двойная, у другого одинарная. Ну а то что они одинаковые это делокализация, все C здесь ароматические и связи у них тоже. B-N контакт в молмехе для BODIPY надо будет отдельно параметризовать.
Кто сейчас на конференции
Сейчас этот форум просматривают: нет зарегистрированных пользователей и 7 гостей