
Прошу покритиковать мою новую статью в блоге про FPD (Feller–Peterson–Dixon):
http://chemcraftprog.com/tmp/fpd.html
Основные тезисы:
- Для проведения высокоточных расчётов нужно принимать во внимание три составляющих: уровень учёта корреляции, размер базисного набора и уровень теории для учёта релятивизма;
- Сейчас в Ab initio преобладает использование подхода Coupled cluster. Этот метод имеет преимущество перед CI – ряды CC сходятся быстрее;
- Метод CCSD(T) называют “золотым стандартом” квантовой химии. В этом методе тройные возбуждения учитываются по теории возмущений, и успешности метода способствует некая компенсация ошибок;
- Для ab initio методов обычно требуется больший базисный набор, так что при использовании метода CCSD(T) не следует ждать “золотого результата” даже с TZ-базисом;
- Композитные методы основаны на аддитивном разложении компонентов энергии;
- Предыдущие композитные методы – G1, G2 – это по-прежнему black box;
- Композитный метод HEAT даёт хорошие результаты, но он требует слишком много ресурсов;
- В статье описана процедура FPD (6 компонентов энергии). Например, формула полной энергии для молекулы ZnSO4:
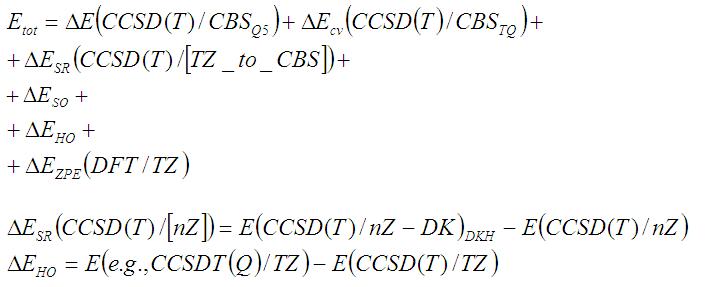
- Есть убедительная гипотеза, что подход FPD никогда не даёт неправильных результатов. Эту фразу надо расшифровать: речь прежде всего о том, что когда квантовик не может достичь сходимости по какому-либо из комонентов, он всегда это знает. Т.е. если метод FPD даёт неточный результат, квантовик это знает;
- Для многих небольших молекул метод FPD позволяет достичь химической, или даже спектроскопической точности, тем более что было показано, что для d и f элементов химической точностью правильнее называть не 1 ккал/моль, а соответственно 3 и 5 ккал/моль;
- Для моделирования спектров часто необходимо убедиться, что работает приближение Борна-Оппенгеймера;
- В одном блоге написано, что для 211 молекул была достигнута химическая точность и на DFT (MAE меньше 1 ккал/моль, MaxAE 2-5 ккал/моль.
