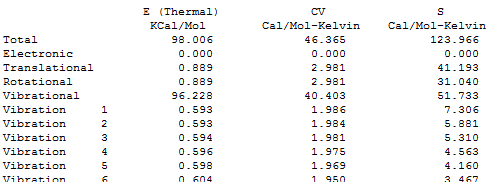
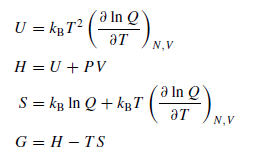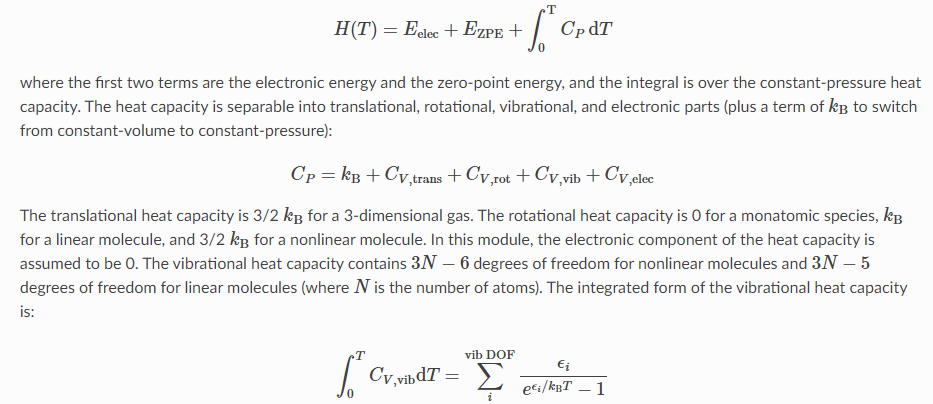Мне нужны книги, где приведены эти формулы. Есть И. Н. Годнев, "Вычисление термодинамических функций по молекулярным данным". Там я нашёл годные формулы для колебательной энтропии и ZPE. Но с формулами для вращательной энтропии, как мне показалось, есть какая-то путаница: я нашёл формулу для молекулы типа симметричного волчка, у которого есть два момента инерции, но не нашёл формулу для несимметричной молекулы с тремя компонентами Ia, Ib, Ic:
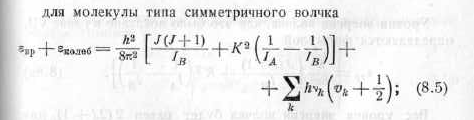
Там далее в книге есть формула для расчёта суммы по состояниям асимметричного волчка, но надо ещё перевести эту сумму по состояниям в энтропию. В связи с этим ещё вопрос - насколько полезно было бы сделать в моей утилите расчёт не только энтропии, ZPE и энергии Гиббса, но и суммы по состояниям? Расскажите, что такое вообще сумма по состояниям и для чего её используют.
Подскажите другие книги с аналогичными формулами. Мне ChatGPT дал такие названия книг, и ничего из этого я не нагуглил:
"Физическая химия" А.И. Китаева и др.;
"Физическая химия: курс лекций" П.В. Балашова и др.;
"Физическая химия. Термодинамика, статистическая физика и кинетика" А.М. Кузнецова и др.