
Ну да две, например атомы водорода в метане.
Расчёт термодинамических функций молекул
Re: Расчёт термодинамических функций молекул
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.
Re: Расчёт термодинамических функций молекул
Про угол C-C-H я не понял, а с этеном действительно, вроде да. Значит для пи-систем нужно вводить какие-то ещё параметры. Могут ли в качестве таких параметров, ну в модели, выступать фиктивные атомы на месте неподелённых электронных пар? Т.е. чтобы этен считался правильно, полагаю стоит добавить четыре фиктивных атома рядом с углеродами, которые тоже отталкиваются и отталкивают другие атомы.Гесс писал(а): ↑Ср авг 09, 2023 2:15 pmТеоретически да, но практически я думаю вы задолбетесь подгонять параметры уже для этана, вам ведь нужно получить не только правильную геометрию но и "квантово-похожий" профиль при малых искажениях. Скажем когда угол C-С-Н отойдет от оптимального. И чем сложнее будет молекула тем ужаснее все будет. Для этилена придется вводить очень большие константы отталкивания, иначе он будет вылетать из плоскости крайне легко.Vit Nhoc писал(а): ↑Ср авг 09, 2023 8:27 amЯ это не понимаю. Разве нужно вводить функционал для углов? Их же много, взаимно зависимых.
Мне казалось, что достаточно ввести функционал для отталкивания между атомами. Например в молекуле этана мы вводим функционал по 7 длинам связей, и плюс к этому отталкивание между всеми парами несвязанных атомов – в результате атомы водорода отталкиваются друг от друга и выстраиваются в (шахматную? В общем D3d) конформацию, в котором двугранный угол H-C-C-H равен 60 градусам. Этого не достаточно?
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.

Re: Расчёт термодинамических функций молекул
По сути вы говорите про замену системы координат длины-углы-диэдралы
на систему длины-длины_между_вторыми_соседями-длины_между_третьими_соседями. Причем последние два это чисто расталкивательный потенциал, видимо асимптотический.
Я видел похожую схему для оптимизации помоему в теракеме (как опцию). Но в силовых полях не сталкивался, впрочем повторюсь в третий раз - я не эксперт в силовых полях.
Но давайте посмотрим что нам светит. Попробуем описать исключительно этан, примем в нем все углы тетраэдральными, связь C-C за 1.5А, C-H за 1.0A. Кроме длин связей вы ставите расталкивающие потенциалы, и думаю немного поиграв с их формой и константами расталкивания C...H и H...H вы сможете заставить углы H-C-H и C-C-H быть тетраэрическими. То есть у вас будет силовое поле способное оптимизировать этан в правильную геометрию, отлично, это первый шаг. Но молмех требует от нас еще и правильную энергетику в геометрии недалеко от минимума.
Начнем с простого - колебание связи С-С. То есть мы сделаем релакс-скан на ДФТ или MP2 по длине этой связи от 1.4 до 1.6 с шагом 0.01А и получим что то похожее на параболу. Теперь нам нужно так подкрутить параметры нашего форсфилда чтоб он воспроизвел эту параболу. Компонент по длине связи даст нам прекрасную параболу и им очень легко манипулировать. А вот расталкивающие потенциалы начнут наклонять параболу (сильно задирать ветвь с "короткой" стороны и мало задирать ее с "длинной"). Это впринципе не так плохо, пока этот эффект мал. То есть нам нужно подкрутить параметры так чтобы воспроизводилась эта парабола и при этом попрежнему правильно считалась геометрия. Думаю это всё еще решаемо, особенно если расталкивающий потенциал зависит от расстояния не просто обратно или обратно квадратно, а по какой то 2-3компонентной формуле.
Но мы идем дальше, просканируем угол C-С-H, опять на релевантном методе, опять получим чтото похожее на параболу (пока мы говорим про отклонения скажем до 10 градусов от оптимального 109). Если бы в нащей модели была пружинка описывающая этот угол - мы бы подкрутили ее силовую постоянную и делу конец. Но у нас расталкивающие потенциалы, причем их подкручивание сразу отразится и на оптимальной геометрии и на описании С-C колебания. Вытянем ли мы это? Я не уверен.
А нам еще предстоит описать вращение метилов (то за что в норме отвечает диэдральный угол), причем воспроизвести в первую очередь барьер этого вращения. И это опять упрется в подкручивание расталкивательных потенциалов. Если мы не лопнули до этого момента - тут я думаю мы таки лопнем. И это всего лишь этан. Слишком много взаимодействий завязано на эти расталкивания, и то что в классической схеме рассовано в сравнительно независимые и легко меняемые параметры (углы и диэдральники каждого типа связей) тут надо переоптимизировать совместо.
на систему длины-длины_между_вторыми_соседями-длины_между_третьими_соседями. Причем последние два это чисто расталкивательный потенциал, видимо асимптотический.
Я видел похожую схему для оптимизации помоему в теракеме (как опцию). Но в силовых полях не сталкивался, впрочем повторюсь в третий раз - я не эксперт в силовых полях.
Но давайте посмотрим что нам светит. Попробуем описать исключительно этан, примем в нем все углы тетраэдральными, связь C-C за 1.5А, C-H за 1.0A. Кроме длин связей вы ставите расталкивающие потенциалы, и думаю немного поиграв с их формой и константами расталкивания C...H и H...H вы сможете заставить углы H-C-H и C-C-H быть тетраэрическими. То есть у вас будет силовое поле способное оптимизировать этан в правильную геометрию, отлично, это первый шаг. Но молмех требует от нас еще и правильную энергетику в геометрии недалеко от минимума.
Начнем с простого - колебание связи С-С. То есть мы сделаем релакс-скан на ДФТ или MP2 по длине этой связи от 1.4 до 1.6 с шагом 0.01А и получим что то похожее на параболу. Теперь нам нужно так подкрутить параметры нашего форсфилда чтоб он воспроизвел эту параболу. Компонент по длине связи даст нам прекрасную параболу и им очень легко манипулировать. А вот расталкивающие потенциалы начнут наклонять параболу (сильно задирать ветвь с "короткой" стороны и мало задирать ее с "длинной"). Это впринципе не так плохо, пока этот эффект мал. То есть нам нужно подкрутить параметры так чтобы воспроизводилась эта парабола и при этом попрежнему правильно считалась геометрия. Думаю это всё еще решаемо, особенно если расталкивающий потенциал зависит от расстояния не просто обратно или обратно квадратно, а по какой то 2-3компонентной формуле.
Но мы идем дальше, просканируем угол C-С-H, опять на релевантном методе, опять получим чтото похожее на параболу (пока мы говорим про отклонения скажем до 10 градусов от оптимального 109). Если бы в нащей модели была пружинка описывающая этот угол - мы бы подкрутили ее силовую постоянную и делу конец. Но у нас расталкивающие потенциалы, причем их подкручивание сразу отразится и на оптимальной геометрии и на описании С-C колебания. Вытянем ли мы это? Я не уверен.
А нам еще предстоит описать вращение метилов (то за что в норме отвечает диэдральный угол), причем воспроизвести в первую очередь барьер этого вращения. И это опять упрется в подкручивание расталкивательных потенциалов. Если мы не лопнули до этого момента - тут я думаю мы таки лопнем. И это всего лишь этан. Слишком много взаимодействий завязано на эти расталкивания, и то что в классической схеме рассовано в сравнительно независимые и легко меняемые параметры (углы и диэдральники каждого типа связей) тут надо переоптимизировать совместо.

Re: Расчёт термодинамических функций молекул
А если нужно описать оптимальную геометрию диметилового эфира? Метанола? Воды?Vit Nhoc писал(а): ↑Ср авг 09, 2023 3:39 pmЗначит для пи-систем нужно вводить какие-то ещё параметры. Могут ли в качестве таких параметров, ну в модели, выступать фиктивные атомы на месте неподелённых электронных пар? Т.е. чтобы этен считался правильно, полагаю стоит добавить четыре фиктивных атома рядом с углеродами, которые тоже отталкиваются и отталкивают другие атомы.
Когда начинает изменять память, практики заводят записную книжку, а романтики садятся писать мемуары.

Re: Расчёт термодинамических функций молекул
Если отталкивание между всеми протонами будет одинаковым, то, не принимая во внимание угловые напряжения, получим "перпендикулярный" этан, как на левой картинкеVit Nhoc писал(а): ↑Ср авг 09, 2023 8:27 amНапример в молекуле этана мы вводим функционал по 7 длинам связей, и плюс к этому отталкивание между всеми парами несвязанных атомов – в результате атомы водорода отталкиваются друг от друга и выстраиваются в (шахматную? В общем D3d) конформацию, в котором двугранный угол H-C-C-H равен 60 градусам. Этого не достаточно?
У вас нет необходимых прав для просмотра вложений в этом сообщении.
Re: Расчёт термодинамических функций молекул
У атома кислорода два таких фиктивных атома.Ahha писал(а): ↑Ср авг 09, 2023 11:20 pmА если нужно описать оптимальную геометрию диметилового эфира? Метанола? Воды?Vit Nhoc писал(а): ↑Ср авг 09, 2023 3:39 pmЗначит для пи-систем нужно вводить какие-то ещё параметры. Могут ли в качестве таких параметров, ну в модели, выступать фиктивные атомы на месте неподелённых электронных пар? Т.е. чтобы этен считался правильно, полагаю стоит добавить четыре фиктивных атома рядом с углеродами, которые тоже отталкиваются и отталкивают другие атомы.
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.
Re: Расчёт термодинамических функций молекул
Почему? Водороды же будут отталкиваться от водородов и углерода противоположной метильной группы.
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.
Re: Расчёт термодинамических функций молекул
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.
Re: Расчёт термодинамических функций молекул
Вернусь к исходной теме. Сейчас столкнулся с дурацкой проблемой: сделал в Chemcraft поднятие малых частот до трешолда, а как обосновать что это правильно - не знаю. Помню что где-то читал про этот подход, но не помню где. Подскажите пожалуйста эту информацию.
Я убедился что с этим подходам лучше согласие с экспериментом, но опубликовать статью мне пока не дают. А ещё нашёл статью Prakhta, где он даёт формулу какого-то другого похожего подхода, возможно более обоснованного, наверно надо будет её закодить тоже. Возможно он называется mRRHO или QRRHO, надо выяснить.
Я убедился что с этим подходам лучше согласие с экспериментом, но опубликовать статью мне пока не дают. А ещё нашёл статью Prakhta, где он даёт формулу какого-то другого похожего подхода, возможно более обоснованного, наверно надо будет её закодить тоже. Возможно он называется mRRHO или QRRHO, надо выяснить.
"Ты должен сделать добро из зла, потому что больше его сделать не из чего". АБ Стругацкие.
Re: Расчёт термодинамических функций молекул
Помню тоже удивился, что теплоемкость мне орка не выдала, выхода нашел два, первый ленивый: просто посчитал для 300К и 301К и разность для внутренней энергии взял, и а второй выход, это колебательную теплоемкость сам считал вставляя значения из орки в производную от энергии колебательных уровней, чуть ли не в экселе(sic!)
У вас нет необходимых прав для просмотра вложений в этом сообщении.
-
some_brero
- Сообщения: 6
- Зарегистрирован: Вс авг 04, 2024 9:20 am
Re: Расчёт термодинамических функций молекул
Тоже считаю вклад колебательных степеней свободы в теплоемкость самостоятельно.
Подскажите, в ORCA можно посчитать энергию возбужденных состояний? На рисунках мои результаты для серы и нескольких молекул. Треугольники соответствуют колебательным частотам, а круги - возбужденным состояниям. Колебательные степени свободы считал сам, а возбужденные состояния брал из справочника. Сравнил с таблицами JANAF.
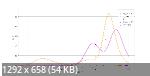

Подскажите, в ORCA можно посчитать энергию возбужденных состояний? На рисунках мои результаты для серы и нескольких молекул. Треугольники соответствуют колебательным частотам, а круги - возбужденным состояниям. Колебательные степени свободы считал сам, а возбужденные состояния брал из справочника. Сравнил с таблицами JANAF.


Кто сейчас на конференции
Сейчас этот форум просматривают: нет зарегистрированных пользователей и 1 гость