выберите первую букву в названии статьи:
А Б В Г Д Е Ж З И К Л М Н О П Р С Т У Ф Х Ц Ч Ш Щ Э Ю Я
Квантовая химия , раздел теоретич. химии, в котором строение и свойства хим. соединений, их взаимод. и превращения в хим. р-циях рассматриваются на основе представлений и с помощью методов квантовой механики. Квантовая химия тесно связана с экспериментально установленными закономерностями в свойствах и поведении хим. соед., в т. ч. с закономерностями, описываемыми классич. теорией хим. строения. Начало развитию квантовой химии положили работы ряда исследователей, выполненные в период становления квантовой механики . В. Гейзенберг (1926) впервые провел расчет атома гелия . В. Гайтлер и Ф. Лондон (1927) на примере молекулы водорода дали квантовомех. интерпретацию ковалентной связи. Их подход нашел дальнейшее развитие в работах Дж. Слейтера (1931) и Л. Полинга (1931) и получил назв. валентных связей метод . В этот же период Ф. Хунд (1928), Р. Малликен (1928), Дж. Леннард-Джонс (1929) и Э. Хюккель (1930) заложили основы широко распространенного в настоящее время молекулярных орбиталей метода . Одновременно появились и основополагающие работы Д. Хартри (1927) и В. А. Фока (1930), создавших самосогласованного поля метод . а также работы Дж. Слейтера (1929-30) по мат. основам конфигурационного взаимодействия метода . X. Бете (1929) и Дж. Ван Флек (1932-35) разработали кристаллического поля теорию . развитие которой привело к созданию поля лигандов теории . нашедшей широкое применение в координац. химии. Общая схема квантовохим. подхода. Квантовохим. рассмотрение атомов, молекул и более сложных систем, свободных или находящихся во внеш. поле, не зависящем от времени, обычно начинается с решения стационарного уравнения Шрёдингера электронов и ядер, входящих в систему. Оператор кинетич. энергии равен:
Notice : Undefined variable: iko in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 95 Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
где индекс i нумерует электроны, индекс a-ядра, т и т a - массы электрона и ядра а соотв., постоянная Планка . В декартовых координатах Di и Da представляют собой сумму вторых частных производных по координатам электрона i и ядра a соотв., например:
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Оператор потенц. энергии равен сумме операторов кулоновского взаимод. частиц, которые зависят от расстояний rij между парами частиц, а также операторов взаимод. частиц с внеш. полем. Напр., для молекулы LiH (4 электрона) в отсутствие внеш. поля гамильтониан выглядит след. образом:
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
При учете спина в гамильтониан входят также операторы спин-орбитального взаимодействия и спин-спинового взаимодействия.
Волновая функция для мол. системы, получаемая в результате решения уравнения Шрёдингера, должна удовлетворять общим требованиям, предъявляемым к волновым функциям в квантовой механике. В частности, для многоэлектронной системы она должна быть антисимметричной относительно перестановки координат (пространственных и спиновых) любой пары электронов, т. е. должна менять знак при такой перестановке (см. Паули принцип ).
Задача нахождения волновой функции и энергии мол. системы обычно решается с помощью вариационного метода или методов возмущений теории . Поскольку соответствующие расчеты весьма трудоемки, в зависимости от сложности объекта и целей исследования используют неэмпирические либо более простые полуэмпирические расчетные методы. В неэмпирических методах заранее задают только числа электронов и ядер в системе, а также заряды ядер и значения фундам. постоянных (напр., постоянной Планка, заряда и массы электрона). В полуэмпирических методах дополнительно из опыта заимствуются значения отдельных входящих в расчет величин, напр, величин, определяющих взаимод. электронов с ядрами, межэлектронное взаимод. и др. (см. Молекулярные интегралы ). При этом вычисляемые величины, например энергия молекулы, взятой в качестве эталона, должны совпадать с эксперим. значениями. Неэмпирич. расчеты, называемые часто ab initio, получили широкое распространение лишь после достаточного развития вычислит. техники и сами в немалой степени способствовали этому развитию.
Обычная схема введения упрощений при решении стационарного уравнения Шрёдингера для молекулы сводится к следующему. В предположении, что центр масс молекулы находится в начале системы координат, вводят адиабатическое приближение . позволяющее решать задачу в два этапа: сначала рассмотреть систему электронов, движущихся в поле неподвижных ядер (электронное уравнение Шрёдингера), а за тем - систему ядер, движущихся в усредненном поле электронов (ядерное уравнение Шрёдингера). На первом этапе тем или иным способом (обычно с помощью прямого вариац. метода) находят волновую функцию системы из N электронов при разл. конфигурациях ядер для k-го электронного состояния Фk (r1 , r2 , ..., rN ; R), где ri (i=1,2,...) - радиусы-векторы электронов, а R - совокупность переменных, определяющих положения ядер (напр., для заданной ядерной конфигурации совокупность независимых расстояний R ab между парами ядер a и b. Одновременно получают и электронные энергии Ek (R), также зависящие от R . Функция Ek (R) определяет тот потенциал, в котором движутся ядра молекулы в ее k-м электронном состоянии. С геом. точки зрения эта функция представляет собой многомерную пов-сть, называемую поверхностью потенциальной энергии или потенц. поверхностью. Функцию Ek (R), вычисленную путем решения электронного уравнения или найденную с помощью к.-л. модельных соображений (напр., в приближении многомерного гармонич. осциллятора), используют на втором этапе, решая волновое уравнение для ядер, предварительно выделив ту часть гамильтониана, которая соответствует вращению системы ядер как целого. Получаемые при этом волновые функции отвечают смещениям ядер друг относительно друга и обычно наз. колебат. волновыми функциями, а соответствующие им собств. значения-колебат. уровнями энергии.
Совр. состояние и перспективы развития квантовая химия Анализ электронного строения молекул (строения электронных оболочек, распределения электронной плотности и др.) позволил интерпретировать разл. типы хим. связей, мн. понятия классич. теории хим. строения и хим. кинетики, такие как валентность . кратность хим. связей, сопряжение и сверхсопряжение, энергия активации хим. реакций и др. На начальных этапах развития квантовая химия были введены в химию новые понятия - гибридизация атомных орбиталей . s- и a-связи, трехцентровые связи, спин-орбитальное взаимод., электроотрицательность атомов, порядки связей, индексы реакц. способности и пр. Были установлены корреляции между вычисляемыми характеристиками (как правило, для равновесных конфигураций атомных ядер молекулы) и свойствами вещества, а также его поведением в хим. реакциях, развита качеств, теория реакционной способности молекул. В 60-е гг. сформулирован и разработан принцип сохранения орбитальной симметрии в хим. реакциях (см. Вудворда-Хофмана правила ). Квантовохим. расчеты поверхности потенц. энергии создали основу для решения задачи об особенностях движения (динамике) ядер частиц, участвующих в элементарном акте хим. реакции. В результате стало возможным вычислять сечения реакции и микроскопич. константы скорости, характеризующие переход из исходного квантового состояния системы в конечное (подробнее см. Динамика элементарного акта хим. реакции).
Благодаря развитию вычислит. методов и значит. совершенствованию неэмпирич. расчетов электронного строения молекул было установлено, что для достижения точности расчетов, сравнимой с точностью лучших эксперим. измерений, вариационные волновые функции должны быть построены на значительно более широком базисе исходных функций, чем при использовании методов мол. орбиталей, и содержать слагаемые, отвечающие не одной, а нескольким разл. конфигурациям электронной оболочки. Это позволяет получить более сложную картину электронного строения молекул, учитывающую локальное, а не усредненное по всем возможным положениям влияние электронов друг на друга, т.е. электронную корреляцию. Расчеты показывают, что во мн. случаях электронная корреляция определяющим образом влияет на энергетические и др. характеристики молекул.
Квантовохим исследования позволили выявить ряд новых особенностей движения ядер частиц, составляющих молекулу. Так, было обнаружено наличие множественных минимумов на потенц. пов-стях, разделенных сравнительно невысокими потенц. барьерами. Кроме того, обнаружена высокая чувствительность электронного строения молекул в возбужденных состояниях к изменению конфигурации их ядер и к малым внеш. возмущениям. Переход к локализованным мол. орбиталям позволил по-новому оценить такие понятия классич. теории хим. строения, как двухцентровые связи, возбуждение той или иной отдельной связи или функц. группы в молекуле и т.п., а также подтвердил возможность использовать характеристики, относящиеся к данному мол. фрагменту (напр., параметры распределения электронной плотности, энергию фрагмента и др.), для всех молекул одного гомологич. ряда, включающих этот фрагмент.
Вычислит. квантовая химия позволяет рассчитывать с достаточно высокой точностью такие важные характеристики молекул, как равновесные межъядерные расстояния и валентные углы, энергии хим. связей, барьеры внутр. вращения и барьеры перехода между разл. конформациями, энергии активации простейших хим. реакций, а также величины, которые затруднительно или даже невозможно определить экспериментально (напр., энергии и геом. параметры молекул в возбужденных состояниях, вероятности квантовых переходов и т.п.).
Квантовая химия позволяет учесть эффекты, связанные с проявлением взаимод. между разл. типами движений в молекулах. Было выяснено, в частности, в каких случаях адиабатич. приближение неприменимо и движение электронов и ядер следует рассматривать одновременно в их взаимодействии (см. Электронно-колебательное взаимодействие ). Такое взаимод. в определенных случаях приводит к неустойчивости симметричной геом. конфигурации молекулы (эффекты Яна-Теллера).
На основе квантовой химии разработана теория электронных спектров поглощения и люминесценции молекул, фотоэлектронных и рентгеноэлектронных спектров. Квантовая теория электрич. и магн. свойств молекул способствовала внедрению в химию физ. методов исследования, в частности ЭПР, ЯМР и ЯКР, и значительно облегчила интерпретацию эксперим. результатов. Получено большое число расчетных данных по вероятностям электронных переходов, временам жизни возбужденных состояний и спектроскопич. постоянным молекул.
К числу осн. направлений развития квантовая химия относятся: всестороннее изучение влияния электронной корреляции на свойства молекул в разл. состояниях и на особенности взаимод. молекул между собой; изучение связи разл. типов движений в молекулах и установление специфики состояний и свойств, в которых эта связь играет определяющую роль (напр., в случае неприменимости адиабатич. приближения); получение и накопление достоверных численных данных высокой точности по свойствам молекул, необходимых для решения прикладных вопросов; развитие теории колебательных и колебательно-вращат. спектров молекул, анализ особенностей колебат. движения при сильном возбуждении многоатомных молекул, переход к локальным колебаниям и др. В исследовании межмолекулярных взаимодействий задачи квантовая химия заключаются в нахождении потенциалов взаимод. при разл. ориентациях молекул, установлении зависимости этих потенциалов от строения молекул, создании моделей, позволяющих учесть влияние среды на свойства молекул и механизмы элементарных процессов. Это позволит решить ряд проблем адсорбции и гетерог. катализа, поведения примесных молекул в твердом теле и др. Разработка этих направлений оказывает заметное влияние на развитие квантовая химия твердого тела.
Изучение химии и физики плазмы, развитие лазерной техники, анализ процессов в атмосфере и космосе потребовали создания новых теоретич. методов, позволяющих исследовать эволюцию мол. систем на основе временного уравнения Шрёдингера. Эти методы применяются, например, при исследовании упругих столкновений атомов, ионов и молекул, развития мол. систем после импульсного их возбуждения лазерным излучением, при анализе динамики элементарного акта хим. реакций, прежде всего газофазных.
Квантовохим. представления и методы начинают активно применяться при изучении высокомол. соед. В частности, созданы адекватные модели для описания высокой проводимости орг. полимеров, переноса заряда по цепи полимера, а также ряда др. процессов. Методы квантовая химия используются в мол. биологии, например для расчета моделей биол. мембран, моделирования работы мышцы и др. Результаты квантовохим. расчетов совместно с данными, получаемыми методами теоретич. физики, начинают использовать в материаловедении для направленного создания материалов с заданными электрич. и магн. свойствами - сплавов, орг. полупроводников, композиц. материалов и др.
Лит.. Цюляке Л., Квантовая химия, пер. с нем., т. 1, М., 1976; Минкин В. И., Сим кия Б. Я., Миняев P.M., Теория строения молекул. Электронные оболочки, М., 1979; Фларри Р., Квантовая химия. Введение, пер. с англ., М., I98S; Минкин В. И., Симкин Б. Я., Миняев Р. М., Квантовая химия органических соединений. Механизмы реакций, М., 1986; Берсукер И. Б., Электронное строение и свойства координационных соединений. Введение в теорию, 3 изд.. Л., 1986; Современные проблемы квантовой химии. Строение и свойства молекул, под ред. М. Г. Вё-селова. Л., 1986; Современные проблемы квантовой химии. Методы квантовой химии в теории межмолекулярных взаимодействий и твердых тел, Л., 1987.
© Н. Ф. Степанов, Н. Д. Соколов.
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice : Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109 Notice : Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
выберите первую букву в названии статьи:
А Б В Г Д Е Ж З И К Л М Н О П Р С Т У Ф Х Ц Ч Ш Щ Э Ю Я
Все новости
Новости компаний
Все новости
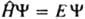 , где E и Y-полная энергия и волновая функция системы,
, где E и Y-полная энергия и волновая функция системы, 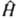 -оператор Гамильтона (гамильтониан) системы, представляющий собой сумму операторов кинетич. и потенц. энергии
-оператор Гамильтона (гамильтониан) системы, представляющий собой сумму операторов кинетич. и потенц. энергии 
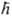 -
-