ЛЮМИНЕСЦЕНЦИЯ (от лат. lumen, род. падеж luminis -свет и -escent - суффикс, означающий слабое действие), свечение вещества, возникающее после поглощения им энергии возбуждения. Представляет собой избыток над тепловым излучением, испускаемым веществом при данной температуре за счет его внутренней (тепловой) энергии. В отличие от др. видов свечения (напр., рассеяния света, тормозного излучения) Л. характеризуется временем свечения, значительно превышающим период колебаний световой волны и составляющим от 10-12 с до неск. суток. Понятие Л. применимо только к такому веществу (совокупности частиц), состояние которого не слишком отличается от термодинамически равновесного, иначе различие между Л. и тепловым излучением теряет смысл.
Механизм Л. заключается в образовании под действием энергии от внеш. или внутр. источника возбужденных состоянийатомов, молекул, кристаллов и послед. испускании ими квантов света (фотонов). По типу возбуждения выделяют фотолюминесценцию (источник энергии возбуждения - свет), радиолюминесценцию (радиоактивное излучение), рентгенолюминесценцию (рентгеновское
излучение), электролюминесценцию (электрич. поле), катодолюминесценцию (пучок электронов), триболюминесценцию (мех. воздействие), хемилюминесценцию (хим. реакции) и др. Различают молекулярную Л., при которой молекулы или атомы испускают фотоны при переходе из возбужденного состояния в основное квантовое состояние, и рекомбинационную Л., когда под действием энергии возбуждения образуются носители заряда (электроны и дырки в кристаллофосфорах) или ионы и радикалы (в газах, жидкостях, стеклах), послед. рекомбинация которых сопровождается испусканием фотонов. Излучат. переход из возбужденного состояния в основное происходит самопроизвольно (спонтанная Л.) или под действием внеш. электромагн. излучения (вынужденная Л.). Испускание света может происходить не обязательно теми же молекулами, которые возбуждаются при поглощении энергии, но и другими, если происходит безызлучат. передача энергии возбуждения (сенсибилизированная Л.). Л. характеризуют спектром испускания (фотолюминесценцию - также спектром возбуждения), квантовым выходом, поляризацией, кинетикой затухания. В данной статье рассматривается мол. фотолюминесценция, которую широко применяют в технике и аналит. химии (см. Люминофоры, Люминесцентный анализ), фотохимии и хим. кинетике для изучения свойств возбужденных состояний частиц и очень быстрых хим. реакций, в фотобиологии, биохимии и медицине для изучения свойств биол. объектов и механизма биол. процессов. О др. видах Л. см. Кристаллофосфоры, Рентгеновская спектроскопия, Хемилюминесценция.
Notice: Undefined variable: iko in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 95
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Механизм Л. Молекулярную фотолюминесценцию подразделяют на флуоресценцию и фосфоресценцию. Флуоресценция характеризуется малой длительностью (менее 10-6 с) и обусловлена испусканием фотонов при переходе системы из возбужденного состояния той же мультиплетности, что и основное состояние. Фосфоресценция -длит. свечение (от долей до неск. десятков с), которое возникает при переходе в осн. состояние из возбужденного состояния иной мультиплетности; такой переход происходит с нарушением спинового правила отбора (см. Квантовые переходы).
Для большинства орг. молекул с четным числом электронов осн. состояние является синглетным, а низшие возбужденные состояния имеют мультиплетность 1 и 3, т. е. могут быть синглетными и триплетными. Для таких молекулфлуоресценция представляет собой излучат. переход в осн. состояние S0 из возбужденного синглетного состояния S1 (переход 2 на рис. 1).
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Рис. 1. Схема квантовых переходов при молекулярной люминесценции. S0 - основной электронный уровень (с колебат. уровнями), S1 и Т1 - возбужденные электронные уровни (синглетный и триплетный соотв.). Прямыми вертикальными стрелками обозначены: поглощение (1), излучательные переходы флуоресценция (2) и фосфоресценция (3), горизонтальными стрелками - безызлучат. переходы: интеркомбинац. конверсия (4) и внутр. конверсия (5). Волнистыми стрелками обозначены процессы колебат. релаксации энергии возбуждения.
С флуоресценцией конкурирует
безызлучат. переход в триплетное состояния Т1 с энергией меньшей, чем у состояния S1 (интеркомбинац. конверсия). Фосфоресценция - излучат. переход из ниж. триплетного состояния Т1 в осн. состояние - наблюдается в условиях, когда конкурирующие с данным излучат. переходом безызлучат. процессы замедлены (высокая вязкость вещества, низкие температуры и т. п.). Впервые связь фосфоресценции с запрещенным излучат. переходом из триплетного состояния была обоснована А. Н. Терениным (1943).
Возможна и т.наз. замедленная флуоресценция, когда вследствие, например, термич. активации молекул в возбужденном триплетном состоянии T1 происходит безызлучат. переход в возбужденное синглетное состояние S1 с
послед. испусканием фотона в результате излучат. перехода S1 : S0. Спектр замедленной флуоресценции идентичен спектру обычной флуоресценции, но время затухания на неск. порядков больше.
У некоторых молекул осн. состояние не является синглетным. Так, для О2 осн. состояние триплетное 3Sg-; слабая фосфоресценция, наблюдаемая для этого вещества в ближней ИК области, обусловлена переходом из ниж. синглетного состояния 1D в основное. Для радикалов, имеющих один неспаренный электрон, осн. состояние дублетное (мультиплетность 2), низшие возбужденные состояния имеют мультиплетность 2 и 4 (соотв. дублетные и квартетные состояния). Флуоресценция радикалов наблюдается при переходе из ниж. возбужденного дублетного состояния в основное.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Спектры Л. Спектр испускания (часто наз. просто спектром Л.) представляет собой зависимость интенсивности свечения от частоты (длины волны) испускаемого света. В лит. обычно приводят наблюдаемые спектры испускания, зависящие от спектральной чувствительности и градуировки прибора. Для получения истинного (квантового) спектра испускания выражают интенсивность Л. в числе фотонов, приходящихся на единичный интервал частот.
Спектром возбуждения Л. наз. зависимость интенсивности свечения на фиксированной частоте от частоты (или длины волны) возбуждающего света. Для получения истинного (квантового) спектра возбуждения необходимо учитывать зависимость интенсивности возбуждающего света (выраженной в числе падающих или поглощенных фотонов в единицу времени) от частоты. При слабом поглощении света образцом истинный спектр возбуждения Л. в большинстве случаев совпадает со спектром поглощения люминесцирующего вещества.
Положение (частота) полосы в спектре Л. определяется разностью энергий состояний, связанных излучательным переходом; интенсивность полосы и время затухания свечения - заселенностью возбужденного состояния и вероятностью перехода (или временем жизни возбужденного состояния). Как правило, Л. происходит при переходе молекулы в осн. состояние S0 с ниж. колебат. уровня первых возбужденных электронных состояний S1 и T1; при возбуждении молекулы в более высокие электронные состояния (S2 и др.) или на верхние колебат. уровни состояний S1 и Т1 избыточная энергия, как правило, релаксирует гораздо быстрее (за время 10-12 с), чем происходит испускание. При испускании фотона сохраняется равновесная ядерная конфигурация молекулы, свойственная возбужденному состоянию (принцип Франка-Кондона), поэтому при возвращении в осн. состояние молекула обычно оказывается на одном из верх. колебат. уровней, соответствующем колебаниям тех хим. связей, равновесная длина которых при данном электронном переходе меняется. В результате в колебат. структуре спектров Л. мн. молекул проявляются частоты колебаний осн. состояния, тогда как в колебат. структуре спектров поглощения проявляются частоты колебаний возбужденного состояния.
В атомных парах и растворах некоторых веществ испускание фотонов происходит при переходе в осн. состояние S0 из того же возбужденного состояния, которое образовалось при поглощении фотона; при этом энергии испускаемого и поглощенного фотонов одинаковы (резонансная Л.); в спектрах испускания и поглощения (возбуждения) наблюдаются совпадающие узкие линии. Чаще, однако, в молекулах, особенно многоатомных, часть поглощенной энергии превращ. в тепловую (происходит диссипация энергии), что приводит к сдвигу спектра испускания относительно спектра возбуждения в низкочастотную (длинноволновую) область (закон Стокса). Спектры высвечивания и возбуждения Л. (имеется в виду наиб. низкочастотная полоса в спектре поглощения) зеркально симметричны относительно прямой, проходящей через точку их пересечения перпендикулярно оси частот, если по оси ординат откладывать интенсивность Л. P(n), выраженную в числе фотонов на единичный интервал частот в единицу времени, или молярный - коэф. поглощения e(n),
а максимумы спектров нормировать к одной и той же величине. Это - т. наз. правило Лёвшина (правило зеркальной симметрии); оно соблюдается в том случае, когда при переходе в возбужденное состояние не происходит существ. изменения мол. структуры и частот колебаний, а меняются лишь равновесные длины связей. Величины P(n) и e(n) связаны между собой универсальным соотношением Степанова:
P(n)/e(n) = Dn3e-hn/kT,
где D - коэф. пропорциональности, не зависящий от n, k и h - постоянные Больцмана и Планка соотв., Т - абс. температура. Если между актами поглощения и испускания фотона существенно меняется структура молекулы в возбужденном состоянии, зеркальная симметрия спектров поглощения и Л. нарушается, и для их описания используют четырехуровневую схему (рис. 2).
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Рис. 2. Четырехуровневая схема квантовых переходов для молекулы, ядерная конфигурация которой в возбужденном состоянии существенно изменяется по сравнению с основным состоянием: 1 - основное состояние (синглетное); 1' - основное состояние с ядерной конфигурацией, соответствующей конфигурации синглетного возбужденного состояния; 2 - возбужденное синглетное состояние с ядерной конфигурацией, соответствующей равновесной конфигурации основного состояния; 2' - возбужденное синглетное состояние с равновесной ядерной конфигурацией (релаксированное).
Как правило, для возбужденных синглетных состояний ji = 1, для триплетных состояний ji [ 1. Если ji не зависит от частоты возбуждающего света, выполняется закон Вавилова, согласно которому квантовый выход Л. постоянен в данной области частот возбуждающего света. Отклонения от закона Вавилова возникают, если при возбуждении в высшие электронные состояния появляются новые пути дезактивации возбужденных молекул, конкурирующие с внутр. конверсией в ниж. возбужденное состояние.
Константу kE можно вычислить из величины момента квантового перехода M21 = <Y2|m|Y1) между двумя электронно-колебательными (вибронными) состояниями, описываемыми волновыми ф-циями Y2 и Y1 (m - оператор дипольного момента):
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
(с - скорость света, п - показатель преломления вещества, n - частота перехода). Экспериментально значения kE в случае флуоресценции определяют из интеграла длинноволновой полосы спектра поглощения:
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
где NA - постоянная Авогадро, - волновое число (см-1). e( ) - молярный десятичный коэф. поглощения (в дм3.моль-1.см-1), < > - среднее значение в спектре флуоресценции:
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
где F( ) - зависимость числа испускаемых фотонов от
волнового числа. Для многоатомных молекул с типичной полушириной полосы поглощения порядка неск. тыс. см-1 справедливо приближенное выражение:
kЕ ~ 104eмакс
(eмакс - молярный десятичный коэф. поглощения в максимуме полосы).
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Кинетика Л. В простых системах мол. Л. после возбуждения коротким (по сравнению с ti) импульсом света затухает обычно по экспоненц. закону: I(t) = I0ехр(-t/ti), где I0 -начальная интенсивность излучения, t - текущее время. Величина, обратная ti, равна сумме констант скорости kj всех излучат. и безызлучат. (включая хим. реакции) процессов гибели данного возбужденного состояния: 1/ti = Sjkj. Для мн. жестких молекул (ароматич. углеводороды, гетeроциклич. соед. и некоторые их производные) ti определяется главным образом константой скорости kISC интеркомбинац. конверсии из состояния S1 в одно из триплетных состояний с меньшей энергией. Величина kISC, в свою очередь, зависит от симметрии электронных волновых ф-ций обоих состояний. Так, для перехода между состояниями одинаковой природы [напр., 1(p, p*) и 3(p, p*)] kISC имеет величину порядка 107-108 с-1, а для состояний разл. природы [напр., 1(p, p*) и 3(n, p*)или 1(n, p*) и 3(p, p*)] она составляет 1010-1011 с-1. В результате молекулы, у которых, например, состояние S1 имеет 1(n, p*) природу, а состояние T13(p, p*) характеризуется меньшей энергией, практически не флуоресцируют, но обладают большим квантовым выходом образования возбужденных триплетных состояний и фосфоресцируют в твердой фазе. У нежестких молекул часто наблюдаются процессы внутр. конверсии , приводящие к релаксации энергии электронного возбуждения и отсутствию как флуоресценции, так и фосфоресценции.
В твердых растворах время жизни молекулы в триплетом состоянии определяется главным образом константами скорости излучат. интеркомбинац. перехода T1 : S0 и безызлучат. электронно-колебат. переноса энергии на сравнительно высокочастотные колебания связей С—Н, О—Н и т. п. в этой же молекуле или в молекуле растворителя. Поэтому квантовый выходфосфоресценции jI лишь в неск. раз меньше квантового выхода jI образования триплетных состояний: jP [ jI = kISCtS, где tS - время жизни состояния S1. В дейтерированных растворителях перенос энергии сильно замедляется и jI приближается к обратной величине константы скорости излучат. интеркомбинац. перехода 1/kP (и может достигать 102 с), а квантовый выходфосфоресценции возрастает. В жидких растворах наблюдается эффективное тушение триплетных возбужденных состояний примесями (в т. ч. растворенным кислородом); их времена жизни падают до 10-5-10-4 с и менее (в зависимости от степени очистки растворителя); при этом фосфоресценция практически исчезает.
При наличии в возбужденном состоянии равновесий между разл. формами возбужденных молекул, адиабатич. реакций или переноса энергии кинетика Л. становится более сложной и м.б. описана суммой двух или неск. экспонент.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Поляризация. Л. обычно частично поляризована даже в случае изотропных образцов и возбуждения неполяризованным светом, если угол между направлениями наблюдения и возбуждения отличен от нуля. наиб. степень поляризации Л. наблюдается в тех случаях, когда направления возбуждения х, наблюдения у и поляризации возбуждающего света z перпендикулярны друг другу, и определяется отношением интенсивностей Iz и Ix компонент Л., поляризованных в направлениях z и х соответственно. Величина Р = (Iz — Iх)/(Iz + Ix) наз. степенью поляризации, а r = (Iz - Ix)/(Iz + 2Ix) = 2P/(3 - P) - анизотропией Л. Поляризация Л. обусловлена анизотропиейдипольных моментов переходов Mij для поглощения и испускания и зависит от угла a между ними по ур-нию Лёвшина- Перрена:
P = (3cos2a - 1)/(cos2a + 3). Для a = 0 Р = 1/2, для a = 90° Р = - 1/3.
Спектр поляризации,
т.е. зависимость степени поляризации или анизотропии от частоты (или длины волны) возбуждающего света, содержит информацию об относит. направленности моментов разл. электронных переходов молекулы. Вращение молекул в маловязких средах и процессы переноса энергии приводят к уменьшению степени поляризации (деполяризация Л.). Влияние вязкости среды h на степень поляризации Л. описывается ур-нием:
1/Р = 1/Р0 + (1/Р0 — 1/3)tkT/vh,
где P0 - эксперим. степень поляризации при очень высокой вязкости среды, v - эффективный объем молекулылюминофора (включая сольватную оболочку), t - время затухания Л.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Тушение Л. Уменьшение квантового выхода наз. тушением Л. Оно может наблюдаться , под действием разл. добавок или примесей (тушителей Л.), при увеличении концентрациилюминофора (концентрац. тушение), при повышении температуры (температурное тушение). Зависимость квантового выхода Л. j от концентрации тушителя [Q] обычно описывается ур-нием Штерна-Фольмера:
j0/j=1+K[Q],
где j0 - квантовый выход Л. в отсутствие тушителя, К -константа тушения. Различают статич. и динамич. тушение Л. Первое обусловлено образованием в осн. электронном состоянии нелюминесцирующих мол. комплексов и не сопровождается изменением времени затухания Л.; при этом К примерно равно константе равновесия комплексообразования (при близких значениях коэф. поглощения комплекса и люминофора на длине волны возбуждения). Второе вызывается взаимод. возбужденных молекул с др. молекулами и сопровождается пропорциональным уменьшением времени затухания (t0/t = j0/j); в этом случае К = kQt0, где kQ - константа скорости такого взаимод., t0 - время затухания Л. в отсутствие тушителя.
О применении Л. в технике и аналит. химии см. Люминофоры, Люминесцентный анализ.
Лит.: Паркер С., Фотолюминесценция растворов, пер. с англ., М., 1972; Лакович Дж. Р., Принципы флуоресцентной спектроскопии, пер. с англ., М., 1986; Modern fluorescence spectroscopy. ed. by E. L Wehry, v. 1-2, L, 1976. М. Г. Кузьмин.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109




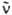 - волновое число (см-1). e(
- волновое число (см-1). e(  ) - молярный десятичный коэф. поглощения (в дм3.моль-1.см-1), <
) - молярный десятичный коэф. поглощения (в дм3.моль-1.см-1), < 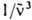 > - среднее значение
> - среднее значение  в спектре
в спектре 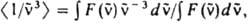
 ) - зависимость числа испускаемых фотонов от
волнового числа. Для многоатомных
) - зависимость числа испускаемых фотонов от
волнового числа. Для многоатомных  , приводящие к
, приводящие к