Notice: Undefined variable: iko in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 95
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
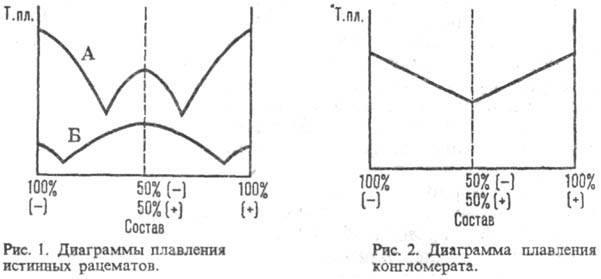
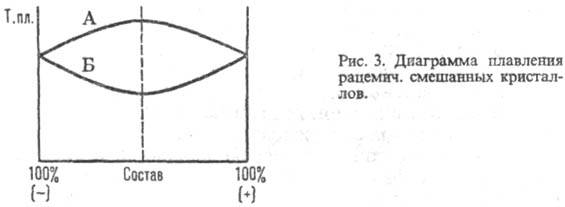
Рацематы, состоят из эквимол. кол-в энантиомеров и не обладают оптич. активностью" Существуют в виде мол. соединений (истинные Р.) и рацемич. смесей кристаллич. энантиомеров (конгломерата, т. е. простой смеси кристаллов право- и левовращающего антиподов) или смешанных кристаллов, образованных обоими энантиомерами. Физ. свойства (т-ра плавления, плотность, растворимость и др.) истинных рацематов отличны от свойств индивидуальных энантиомеров, а их ИК спектры и рентгенограммы отличаются от тех, которые дают простые смеси этих же веществ. Образование истинных рацематов обусловлено водородными связями, индукционным или дисперсионным взаимодействием. Характер связи между энантиомерами в рацематах может быть определен с помощью диаграммы зависимости температуры плавления от состава: для истинных рацематов (рис. I) она имеет эвтектич. точки и максимум, соответствующий соотношению энантиомеров 1:1, температура плавления м. б. как выше (линия Б), так и ниже (А) температур плавленияэнантиомеров; для конгломерата - резкий минимум в точке эквивалентности (рис. 2); для смешанных кристаллов диаграмма м. б. выпуклой (А), вогнутой (Б) или прямой линией (рис. 3).
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Образование молекулярных соед. возможно также при смешении энантиомерных форм родственных соед., например (+)-хлорянтарной и (—)-бромянтарной кислот. Подобные раце-мич. соед. наз. квазирацематами. Их диаграммы плавления сходны с диаграммами истинных рацематов, но обе половины кривой состояния уже не симметричны и максимум может и не соответствовать энантиомерному составу 1:1. Образование квазирацематов используют для определения конфигурации молекул (метод квазирацематов). Метод заключается в том, что по характеру диаграммы плавления смеси двух веществ определяют, являются ли они энантиомерами или нет, и, если конфигурация молекул одного из веществ известна, устанавливают конфигурацию молекул второго. Об образовании квазирацематов можно судить также по ИК спектрам и рентгенограммам, которые, как и у истинных рацематов, отличны от спектров и рентгенограмм простых смесей двух веществ.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Рацематы образуются при любом хим. синтезе, приводящем к хиральным молекулам, если исходные компоненты реакции были оптически неактивны и синтез проводился в отсутствие асимметризующих воздействий (хиральный катализатор, облучение циркулярно-поляризованным светом и др.; см. Асимметрический синтез). Это обусловлено тем, что переходные состояния при образовании энантиомеров энергетически эквивалентны. При наличии асимметризующих факторов переходные состояния диастереомерны, их энергия различна, поэтому возможно образование предпочтительно одного из двух энантиомеров конечного продукта.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Рацемизация. Рацематы образуются также в результате рацемизации оптически активных соед., представляющей собой обратимое взаимное превращение энантиомеров. В отсутствие асимметризующих факторов этот процесс заканчивается установлением динамич. равновесия между ними при строго эквимолярном содержании энантиомеров в смеси.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
При рацемизации происходит обмен местами к.-л. двух атомов или радикалов, связанных с элементом хиральности. Рацемизация - часто не самопроизвольный процесс; она вызывается, например, действием кислот, щелочей, повышением температуры.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Скорость и механизм рацемизации зависят от строения оптически активных соед. и от условий ее проведения (т-ры, растворителя, катализатора и т.д.). Кинетически рацемизация обычно описывается ур-нием для необратимой реакции первого порядка:
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
k= (2,3/t)lga0/at,
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
где k-константа скорости рацемизации, a0 и at-величины соотв. первонач. оптич. вращения и ко времени t. В зависимости от природы элементов хиральностимолекулы энан-тиомера рацемизация м. б. либо химической, либо физической.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Хим. рацемизация наиб. характерна для соед., в молекулах которых хиральным центром является асим. атомуглерода. Отрыв от него одного из заместителей приводит к образованию плоского карбкатиона и потере хиральности. Послед. присоединение этого же заместителя происходит равновероятно с обеих сторон плоскости карбкатиона, что приводит к образованию рацематов. По др. механизму рацемизация протекает с промежут. образованием карбаниона, например в результате отрыва протона от асим. атома С. При термич. рацемизации в результате гомолитич. разрыва связи асим. атома с одним из заместителей образуются радикалы, которые при рекомбинации дают рацематы.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Легкость протекания рацемизации зависит от типа функц. групп, связанных с асим. атомом С. Легко рацемизуются соед., содержащие в качестве заместителей при асим. центре атомводорода и сильный акцептор электронов, напр, молочная кислота СН3—СНОН—СООН, дикетоны R—СО— —CHR'—COR: и т.д. У дикетонов хиралъность исчезает в результате енолизации. В то же время соед., не склонные к образованию промежут. ионов или таутомерным превращениям, например алканы, устойчивы к рацемизации. Существуют соед., которые хотя и образуют промежут. ионы, не подвергаются рацемизации, вследствие стерич. особенностей структуры их молекул. Напр., у производных камфоры или триптицена, в молекулах которых асим. атом С находится в вершине циклич. системы, присоединение протона к промежут. карбаниону возможно только со стороны, противоположной циклич. системе:
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Физ. рацемизацию наиб. легко проследить на примерах таких родственных соед., как амины, фосфины, арсины, стибины 3R1R2R3. Молекулы этих соед. имеют неподеленную паруэлектронов, и при наличии разл. заместителей у гетероатома для них возможна оптич. изомерия. Рацемизация знантиомерных форм этих соед. обусловлена пирамидальной инверсией. В случае аминов вследствие быстрой инверсии выделить оптич. изомеры обычно не удается; амины существуют только в виде рацематов. Исключения - циклич. соед. (основания Трегера, азиридины, диазиридины) и третичные амины NR1R2R3, содержащие в радикалах R элект-роотрицат. заместители. С ростом размера гетероатома в ряду N < Р < As < Sb величина энергетич. барьера инверсии для соед. 3R1R2R3 возрастает, соотв. увеличивается и устойчивость этих соед. к рацемизации. Стибины вполне стабильны и при нормальных условиях могут существовать в виде рацематов и отдельных энантиомеров.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Без к.-л. хим. реакций происходит рацемизация соед. с мол. асимметрией. Такие соед. рацемизуются в результате взаимного перемещения (гелицены) или вращения (производные дифенила) отдельных фрагментов их молекул. Если энергетич. барьер этих перемещений достаточно высок, соед. устойчивы к рацемизации (производные дифенила с четырьмя объемистыми заместителями в орто-положениях или гелицены, содержащие в молекуле более шести конденсир. колец), в случае же малых энергетич. барьеров рацемизация осуществляется достаточно легко.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Расщепление рацематов. Обратный рацемизации процесс - выделение энантиомеров из их рацемич. смеси - наз. расщеплением рацематов. Впервые расщепление рацематов было осуществлено (Л. Пастер, 1848) при кристаллизации натрий-аммониевой соли виноградной кислоты; выделенный осадок представлял собой энантиоморфную смесь кристаллов, а индивидуальные кристаллы-либо лево-, либо правовращающие формы винной кислоты. Известно лишь неск. десятков примеров расщепления рацематов при спонтанной кристаллизацииэнантиомеров. Более общий метод заключается в том, что в пересыщ. раствор рацемата вводят затравку кристаллов одного из энантиомеров, что приводит к кристаллизации именно этого оптич. изомера. Затем в оставшийся раствор добавляют затравку кристаллов второго энантиомера и тем самым вызывают его кристаллизацию, поскольку именно этим оптич. изомером пересыщен оставшийся раствор, и т.д. Расщепление Р. путем затравочной кристаллизации реализовано в промышленности (напр., для D,L-глутаминовой кислоты), однако этот способ также не универсален.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Др. способ расщепления Р.-биохимический-основан на том, что микроорганизмы при своем развитии используют только один из двух оптич. изомеров, присутствующих в Р. Остающийся энантиомер м. б. выделен. Этот путь позволяет получать только один из энантиомеров, второй необратимо теряется. Избирательность действия микроорганизмов по отношению к энантиомерам связана с высокой энантиоселективностью содержащихся в микроорганизмах ферментов. Поэтому для разделения энантиомеров нет необходимости применять сами микроорганизмы, достаточно использовать в этих целях выделенные из биол. объектов ферментные препараты. Наиб. широко для расщепления рацематов применяют гидролазы - ферменты, катализирующие гидролиз сложноэфирных или амидных связей. При этом гидролизу подвергается только один из двух энантиомеров субстрата, а разделение конечной смеси, например, своб. кислоты и ее сложного эфира м. б. легко осуществлено обычными методами. Так, при действии фермента ацилазы на рацемич. N-ациламинокислоту гидролизу (а следовательно, и отделению) подвергается лишь L-форма.
Notice: Undefined variable: goo in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
Notice: Undefined variable: mn in /var/www/chemport.ru/data/www/chemport.ru/chemical_encyclopedia.php on line 109
В то же время высокая специфичность действия ферментов ограничивает их использование, поскольку мн. синтетические рацематы, не встречающиеся в живой природе, не подвергаются воздействию ферментов. Др. недостаток этого метода-относительно высокая стоимость ферментов. Тем не менее расщепление рацематов с использованием иммобилизованных на нерастворимом носителеферментов реализовано в пром. произ-ве оптически активных аминокислот.
Наиб. общий метод расщепления Р.-химический, при котором на Р. действуют оптически активным реагентом, в результате чего образуется новая пара веществ - диастереоме-ров. Последние м. б. разделены вследствие различия в их физ. свойствах. Хиральный реагент после разделения диасте-реомеров отщепляют. Напр., рацемич. (R, S)-1-фенилэтил-амин образует с природной (2R, 3R)-винной кислотой две диасте-реомерные соли: [(R)-1-фенилэтиламин]·[(2R,3R)-винная кислота] и [(S)-1-фенилэтиламин]·[(2R, 3R)-винная кислота], которые обладают разл. растворимостью в этаноле и м. б. разделены кристаллизацией. Своб. амин выделяют затем экстракцией диэтиловым эфиром из водного щелочного раствора соли.
Для расщепления рацемич. кислот используют их способность образовывать соли с хиральными прир. и синтетич. основаниями-хинином, бруцином, стрихнином, 1-фенилэтиламином и др. Рацемич. спирты расщепляют путем превращения их в кислые эфиры дикарбоновых кислот (напр., фталевой) с послед. кристаллизацией в виде диастереомерной соли с хиральным амином.
Метод расщепления Р. путем превращения их в диастерео-меры не пригоден для орг. соед., не имеющих функц. групп, например для алканов. Для расщепления таких Р. используют, например, способность мочевины к образованию клатратов. Мочевина кристаллизуется в хиральной гексагон. решетке, в цилиндрич. каналах которой могут размещаться молекулы "гостя". Кристаллымочевины м. б. как право-, так и левовращающими, поэтому клатратымочевины с энантиомерами приобретают характер диастереомеров. Разделение Р. по этому способу осуществляют внесением затравки одного из энантиомеров в насыщ. раствор мочевины и Р., выпадающие при этом кристаллыклатрата обогащены именно внесенным энантиомером.
Др. хим. метод расщепления рацематов - кинетич. расщепление, основанное на том, что в реакциях с оптически активными реагентами (или в присутствии хиральных катализаторов или хиральных растворителей) скорость превращения одного энантиомера не равна скорости превращения другого. Если в подобную реакцию ввести рацемат и прервать реакцию до ее полного завершения, то один из энантиомеров, реагируя быстрее, будет преобладать в продукте реакции, другой-в непрореагировавшем остатке. Пример - расщепление рацемич. (n-толил) мезитилсульфоксида восстановлением под действием реагента, полученного из оптически активного 1-фенилэтиламина и А1Н3.
Асим. превращениями наз. процессы, в ходе которых происходит превращение рацематов в смесь энантиомеров с преобладанием одного из них. К асим. превращениям относится, в частности, мутаротациямоносахаридов. Этот метод наз. также ретрорацемизацией.
Для расщепления рацематов используют также хроматографию на хиральных неподвижных фазах. Ранее в качестве таких фаз использовали прир. хиральные полимеры - белки, крахмал, целлюлозу, шерсть и др., из которых теперь применяют только микрокристаллич. целлюлозу и ее производные. В осн. для расщепления рацематов используют более селективные синтетич. хиральные сорбенты, полученные специально для разделения той или иной группы рацемич. соединений. Так, Р., молекулы которых содержат фрагменты, способные к образованию комплексов с переносом заряда, м. б. расщеплены на хиральных фазах, имеющих структуру нафтилглицина (для электроноакцепторных молекул) или динитробензоил-аминокислот и пикрил (1-фенилэтил) амина (для электроно-донорных молекул). Соед., способные к образованию комплексов с переходными металлами, м. б. разделены на энантиомеры методом лигандообменной хроматографии с использованием хиральных комплексообразующих сорбентов. Этим методом расщепляют, например, меченные тритием a-аминокислоты. Применяют сорбенты, содержащие фрагменты циклодекстринов, расщепление рацематов на них осуществляется в результате образования соед. включения. Получены хиральные стационарные фазы, разделение энантиомеров на которых происходит благодаря возникновению водородных связей и ион-дипольных взаимод. между энантиомерами и сорбентом. Расщепление рацематов хроматографич. методом позволяет одновременно получить и информацию об оптич. чистоте выделенных энантиомеров.
Лит.: Потапов В.М., Стереохимия, 2 изд., М., 1988; Jacques J., Collet A., Wilen S.H., Enantiomers, racemates and resolutions, N. Y., 1981.